





En 2016 el brote del virus del Zika (ZIKV) tuvo una fuerte repercusión debido a la cantidad de casos observados, especialmente en Brasil. Sorprendentemente, aún no se conoce mucho acerca de la epidemiología de la enfermedad causada por ZIKV, en parte por la escasa información genómica disponible.
Un grupo de investigadores de la Fundación Oswaldo Cruz (Fiocruz) de Río de Janeiro, en colaboración con el Instituto Broad del Massachusetts Institute of Technology (MIT) y de la Universidad de Harvard, llegaron a un importante descubrimiento sobre la entrada del virus del Zika a las Américas.
Sus hallazgos, que incluyen evidencias sobre la ruta del virus, fueron publicados en la prestigiosa revista Nature en mayo de este año. Para interiorizarnos sobre la investigación y sobre la situación actual de ZIKV, Ciencia del Sur conversó con uno de los autores de la publicación, el Dr. Edson Delatorre, investigador de la Fiocruz.
Edson Delatorre realizó su doctorado en Biología Parasitaria (virología) en la Fiocruz, en el Laboratorio de SIDA e Inmunología Molecular. Su tesis doctoral versó sobre la evaluación de la prevalencia de mutaciones transmitidas de resistencia a los antirretrovirales y de la historia evolutiva de agrupaciones raras de HIV-1 en Río de Janeiro, en una población de gestantes antes del inicio de la terapia para prevención de la transmisión vertical.
Actualmente es investigador posdoctoral en el mismo instituto y se dedica al estudio de los mecanismos que rigen la dinámica evolutiva viral intra e interhuésped, con enfoque en filogeografia y filodinámica.
Sobre a la situación de la enfermedad en Brasil, expresó lo siguiente: “Luego de la explosión de casos probables de fiebre causada por el virus del Zika en Brasil en 2016, observamos una reducción drástica en el número de casos probables en 2017. Esa reducción se debería al agotamiento de la población susceptible con el avance de la enfermedad, ya que el virus posee solamente un serotipo, y de esta forma el individuo se vuelve inmune después de la infección, incluso si ésta fue asintomática”.
Para el científico, esa dinámica es diferente de la observada para el virus Dengue (DENV) que posee cuatro serotipos. En el caso de DENV, ocurre una alternancia de serotipos que circulan y que cambian a medida que la población se vuelve resistente a un determinado serotipo.
“Lo que podemos esperar en los próximos años con relación al virus del Zika es el retorno de la infección cuando la población de niños (nacidos después de este brote y por lo tanto aun susceptibles) se acumule a niveles suficientemente grandes para sostener una nueva ola de transmisión. El monitoreo continuo de este virus, como también el desarrollo y mejoramiento de kits que permitan el diagnóstico diferencial con otras arbovirosis con síntomas semejantes, como dengue y chikunguña, son de importancia fundamental para la identificación precoz de esta posible nueva epidemia para que acciones de control sean realizadas”, señaló el especialista.
Siguiendo al virus
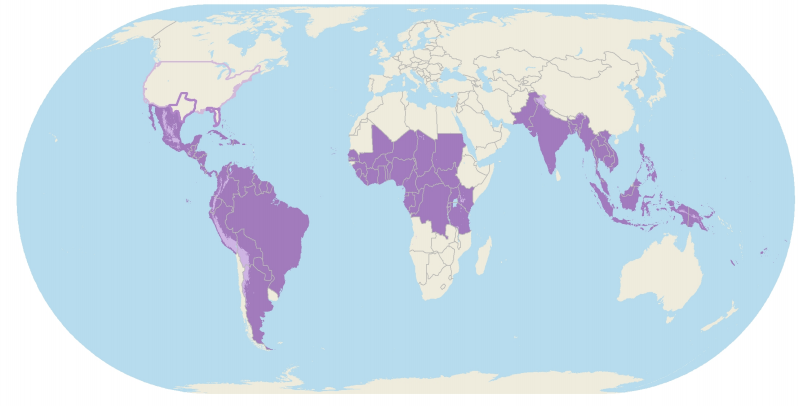
Uno de los objetivos principales del trabajo que realizaron fue conocer la trayectoria del virus. Con respecto a este punto y sobre la importancia del mismo, Delatorre comentó: “Una vez que se conozca a partir de dónde y cuándo una epidemia se estableció, es posible intentar estimar cuál será el próximo paso que dará. En este caso, la importancia de este conocimiento no se restringe solamente al virus del Zika, sino también a otros virus de gran importancia epidemiológica, como la influenza y el ébola. No siempre es posible reconstruir la trayectoria del virus en forma directa utilizando solamente datos epidemiológicos”.
“Esta es una de las principales limitaciones de los estudios con el virus del Zika, ya que la mayor parte de los casos puede ser asintomática. Para eso, estudios de evolución viral, combinando informaciones genómicas y epidemiológicas pueden ayudar a elucidar cómo una epidemia se dispersa en el tiempo y en el espacio, posibilitando construir modelos que ayuden a entender cómo ocurrieron las transmisiones y, en el caso del trabajo que publicamos, cuáles fueron los caminos de diseminación del ZIKV en las Américas”, añadió el científico.
El ZIKV tiene al ARN (Ácido Ribonucleico) como material genético en lugar del ADN (Ácido Desoxirribonucleico) y es sabido que los virus ARN poseen alta diversidad genética. “A pesar de presentar una diversidad grande, la misma se encuentra dentro del intervalo observado en otros virus RNA. Normalmente, cuando ocurre la emergencia de un nuevo virus (como ocurrió en el caso de Ébola en África Occidental y ahora con el ZIKV), las personas se asustan porque escuchan la palabra mutación, imaginándose que nuevos virus mutantes van a surgir, ganando nuevas formas de transmisión o causando nuevos síntomas”, expresó el investigador.
Delatorre apuntó que las mutaciones en realidad ocurren todo el tiempo durante la replicación viral y es natural que ocurran más cuando un virus se establece en una nueva población, que por ser altamente susceptible, favorece a que más ciclos replicativos ocurran.
“Esto no quiere decir que sea imposible que ocurra lo que las personas piensan, al final, mutaciones son la base de la evolución. Lo que se debe tener en mente es que la probabilidad de que estos eventos ocurran es baja, una vez que la mayor parte de las mutaciones terminan siendo deletéreas, no proporcionando ninguna ventaja al virus”, aseguró.
Una parte importante de la metodología aplicada en el trabajo publicado en Nature, fue la utilización de muestras clínicas secuenciadas directamente sin cultivo previo. “Lo que se anhela al realizar el cultivo viral es amplificar la cantidad de virus, ya que el número de partículas virales presente en las muestras muchas veces es muy pequeño para ser secuenciada directamente. En este caso, la amplificación viral es realizada después de la replicación. Esto puede acarrear el aparecimiento de selección de mutaciones”, comentó.
“De esta forma, al secuenciar virus oriundos de cultivo, la secuencia generada probablemente será diferente del genoma aislado originalmente, lo que puede traer consigo errores en los análisis subsecuentes. Por eso elegimos la realización del secuenciamiento directo de las muestra para así obtener genomas lo más próximo posible a los genomas reales de los aislados virales”, agregó.
Problemas con el ARN
El ARN representa comúnmente desafíos en el laboratorio comparado con el ADN. Por ejemplo, el ARN tiene mayor facilidad de contaminación, inestabilidad molecular, entre otros.
En relación a las dificultades encontradas al trabajar con el ZIKV, tanto en el laboratorio como en el análisis bioinformático, Delatorre nos contó: “Una de las dificultades es la viremia efímera y en bajos niveles en los individuos infectados, lo que vuelve el secuenciamiento directo de la muestra clínica mucho más difícil. Este es uno de los principales motivos de la baja cantidad de genomas disponibles de este virus, y por eso, nuestro trabajo presentó una gran contribución al generar más de 100 genomas virales de muestras clínicas humanas y de mosquitos de 10 países y territorios americanos”.
Finalmente, le consultamos sobre los desafíos posteriores que genera este avance: “Una de las primeras preguntas que surgen es ‘¿por qué ahora?’. Después de circular por más de 50 años en Asia causando casos esporádicos, este virus comenzó a causar brotes aislados a partir de la segunda mitad de la década de 2000, pero solamente alrededor de 2014 fue introducido en América. ¿Por qué eso no sucedió antes? ¿Será que fue causado por algún factor genético que favoreció la dispersión del virus? Algunos indicios apuntan que sí”.
“Recientemente, una investigación demostró que aislados virales del brote americano eran más infecciosos en mosquitos que aislados asiáticos, aparentemente debido a una única mutación en la proteína NS1. Intentar reconstruir mejor los pasos evolutivos ocurridos entre la diseminación del ZIKV desde Asia para las Américas será esencial para intentar responder estas preguntas», indicó.
«Otra cuestión importante es la de cuándo y dónde ocurrirá la próxima epidemia del ZIKV, tanto en el Brasil como en otros países americanos. Solamente con el seguimiento continuo y con el desarrollo de mejores tests de diagnóstico podremos prepararnos para cuando este momento llegue”, concluyó el científico.
Referencia
- Hayden C. Metsky, Christian B. Matranga, Shirlee Wohl, Stephen F. Schaffner, Catherine A. Freije, Sarah M. Winnicki, et al. Zika virus evolution and spread in the Americas. Nature 546, 411–415. 2017. doi:10.1038/nature22402
¿Qué te pareció este artículo?

 (9 votos, promedio: 4,33 de 5)
(9 votos, promedio: 4,33 de 5)Columnista y editora científica de Ciencia del Sur. MSc y PhD en Biología Parasitaria con énfasis en Biología Molecular aplicada a microorganismos por el Instituto Osvaldo Cruz (Fiocruz) de Río de Janeiro, Brasil. Fabiola obtuvo su licenciatura en Biología de la Facultad de Ciencias Naturales y Exactas de la Universidad Nacional de Asunción.
Realizó un posdoctorado en la Universidad de Bath (Inglaterra) y es colaboradora externa del Centro para el Desarrollo de la Investigación Cientifica. Fue Research Assistant en el Instituto Sanger de Cambridge. Actualmente, es Senior research technician in NGS. Departamento de Oncología de la Universidad de Cambridge, Inglaterra.
- Fabiola Román Maldonado

- Fabiola Román Maldonado
- Fabiola Román Maldonado
- Fabiola Román Maldonado
- Fabiola Román Maldonado
- Fabiola Román Maldonado
- Fabiola Román Maldonado
- Fabiola Román Maldonado
- Fabiola Román Maldonado
- Fabiola Román Maldonado
- Fabiola Román Maldonado
- Fabiola Román Maldonado
- Fabiola Román Maldonado
- Fabiola Román Maldonado
- Fabiola Román Maldonado
- Fabiola Román Maldonado
- Fabiola Román Maldonado
- Fabiola Román Maldonado
- Fabiola Román Maldonado
- Fabiola Román Maldonado
- Fabiola Román Maldonado
- Fabiola Román Maldonado
- Fabiola Román Maldonado
- Fabiola Román Maldonado
- Fabiola Román Maldonado
- Fabiola Román Maldonado
- Fabiola Román Maldonado
- Fabiola Román Maldonado
- Fabiola Román Maldonado
- Fabiola Román Maldonado
- Fabiola Román Maldonado






























Excelente artículo. Debemos estar preparados para la segunda oleada del Zika, con un sistema de vigilancia epidemiologica sensible y un robusto y eficiente equipo de respuesta. Esta investigación oportunamente realizada por el investigador nos muestra la relevancia de hacer ciencia sobre los problemas que nos aquejan como región.