






La aplicación, adaptación y el desarrollo de nuevas de técnicas laboratoriales avanzadas para la vigilancia epidemiológica nacional es una necesidad impostergable que se ha vuelto aún más urgente en la actual pandemia.
Dicho emprendimiento científico, además de responder a los desafíos de salud pública e integrar la investigación con la vigilancia, asistirá fuertemente al posicionamiento del país entre los actores científicos internacionales.
Mediante el apoyo del Consejo Nacional de Ciencia y Tecnología (CONACYT) a través del Programa PROCIENCIA, financiado por el Fondo para la Excelencia de la Educación y la Investigación (FEEI), investigadores paraguayos se alistan para la secuenciación del genoma completo del SARS-CoV-2, virus causante de la COVID-19.
La propuesta, que fue adjudicada por el CONACYT dentro de la convocatoria “Proyectos de Investigación en el Contexto de la Pandemia del COVID-19”, debe iniciar el procesamiento de muestras en breve.
El proyecto, denominado «Estudio de la dinámica de transmisión y de la variabilidad genética de SARS-CoV-2 circulantes en Paraguay a través del análisis de secuencias del genoma viral», tiene como objetivo obtener secuencias completas de genoma de SARS-CoV-2 aislado de casos nacionales de COVID-19.
El trabajo contempla la obtención y el análisis de 100 genomas de SARS-CoV-2 circulantes en Paraguay en una primera etapa, resultado de la implementación de los 3 pasos fundamentales de la metodología: la técnica de secuenciación (que será la técnica por nanoporos), el procesamiento bioinformático de la información arrojada por el secuenciador y el análisis filogenético de los genomas obtenidos.
Será ejecutado por el Centro de Estudios y Formación para el Ecodesarrollo – ALTER VIDA e investigadores de la Universidad Nacional de Asunción (1), quienes están en proceso de adquirir los equipos y reactivos necesarios para la investigación. Las adquisiciones además servirán a futuro para la realización de otros proyectos similares, incluyendo otras aplicaciones de vigilancia de la salud.
Entre los objetivos se encuentran el análisis del genoma viral completo, la realización de un análisis del grado de variabilidad de los distintos genes del virus que circula en el país, así como el estudio de cómo este se relaciona con las variantes circulantes alrededor del mundo.
Al estudiar las variaciones que presenta el genoma de los virus analizados, será posible estimar el patrón de diseminación en el tiempo de las variantes desde el inicio de la circulación viral en nuestro país. Además, se podrán identificar las posibles variantes circulantes actualmente en parte del territorio nacional y cómo se comportan las mismas en nuestra población.
De acuerdo al portal del CONACYT, otros objetivos del trabajo son estimar el tiempo de introducción del virus al país y la tasa de crecimiento de casos en base a la tasa mutacional, lo que aportará datos sobre la biología de este virus pandémico. (1)
Los investigadores que están envueltos en la aplicación de este proyecto tienen amplia experiencia en el área de la virología, la biología molecular y genética, y han trabajado en la detección del virus mediante la técnica de RT-PCR en Paraguay.
Recordemos que la primera secuencia del genoma completo del SARS-CoV-2 se obtuvo en Wuhan, China, a finales de diciembre de 2019 y permitió identificar al virus como un coronavirus y comprobar que era diferente a los otros ya conocidos.
¿Qué es la secuenciación del ADN y por qué es tan importante?
Primeramente, debemos clarificar que la secuenciación de una muestra de DNA es un proceso diferente a la técnica de reacción en cadena de la polimerasa o PCR. La complejidad de ambos procesos es alta. Por tanto, aquí solo aspiramos a dar una visión general.
Durante el 2020, la técnica de PCR ha pasado a ser muy conocida por el público general debido a su utilización para la detección de SARS-CoV-2 en muestras de hisopado nasofaríngeo. Si bien la PCR se basa también en la manipulación del material genético del virus (si está presente), la misma recurre solo a la identificación de un fragmento relativamente pequeño del genoma. Esta es la principal diferencia.
Luego de una extracción de material genético (ARN O ADN), la técnica de PCR sirve para amplificar un fragmento específico de ADN (i.e. aumentar el número de sus copias), siempre que este se encuentre presente en la muestra estudiada.
El principal beneficio de esto es que, tras la amplificación, resulta mucho más sencillo identificar (con sensibilidad y especificidad muy altas) microorganismos causantes de enfermedades, identificar personas o hacer investigación científica sobre el ADN amplificado.
Además, la PCR tiene varias adaptaciones, lo que facilita la detección del material genético de distintos organismos.
La secuenciación del genoma completo, por otro lado, significa determinar el orden de los cuatro componentes químicos básicos, llamados bases nitrogenadas, que forman la molécula de ADN. Con este análisis se consigue acceder a la información genética de un organismo.
Dependiendo del objetivo de investigación, se pueden secuenciar desde fragmentos de ADN hasta el genoma completo de organismos, como el estudio en cuestión propone hacer con el SARS-CoV-2. La secuenciación de ADN ha pasado a ocupar un lugar trascendental en el estudio de los diferentes organismos que existen en la Tierra.
Desde la finalización del Proyecto Genoma Humano, en el 2003, el avance de la tecnología ha hecho que genes individuales puedan secuenciarse rutinariamente en muchos laboratorios en un tiempo corto y con un bajo costo (comparado con los inicios de la secuenciación) (2).
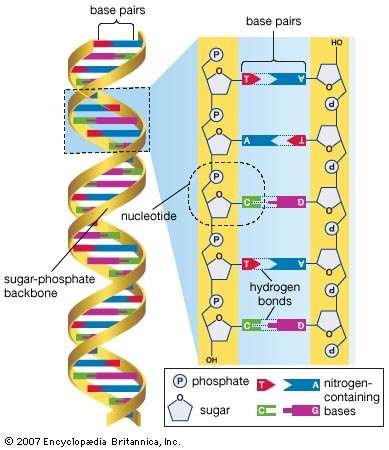
Los nucleótidos son los peldaños que componen el ADN y están formados químicamente por un fosfato, un azúcar (la desoxirribosa) y las bases nitrogenadas. En una molécula de ADN (cuya estructura es una doble hélice), las bases nitrogenadas presentes son: Adenina, Guanina, Citosina y Timina. (Figura 1)
En la doble hélice de ADN, las cuatro bases químicas se unen con la misma pareja para formar «pares de bases». Adenina (A) siempre forma pareja con timina (T); citosina (C) siempre forma pareja con guanina (G). Es importante señalar que existen otros tipos de emparejamientos que pueden surgir, pero estos escapan del alcance de la presente discusión.
El orden de los ácidos nucleicos en las cadenas de polinucleótidos contiene, en última instancia, la información de las propiedades hereditarias y bioquímicas de la vida. Por lo tanto, la capacidad de medir o inferir tales secuencias es imprescindible para la investigación biológica.
La secuencia informa a los científicos la clase de información genética que se transporta en un segmento específico de ADN (2).
Secuenciar un genoma completo, es decir, acceder a la información genética completa de un organismo, sigue siendo una tarea compleja. El proceso requiere romper el ADN del genoma en muchos pedazos más pequeños, secuenciar dichos pedazos y ensamblar las secuencias en una única y larga «secuencia consenso”.
Dos generaciones de técnicas de secuenciación
Las técnicas de secuenciación han mejorado con el correr de los años. Inicialmente los análisis se realizaban de manera manual, incluyendo las técnicas en el laboratorio, lo cual, hacia el proceso muy lento y laborioso, muy laborioso.
Una de las primeras técnicas de secuenciación fue desarrollada por Allan Maxam y Walter Gilbert (secuenciamiento químico) (3). Este método no utilizaba programas bioinformáticos y se basaba en el análisis de geles de poliacrilamida para visualizar cuál nucleótido era identificado.
Los descubridores del método habían reportado con esta técnica la secuenciación de unos 24 pares de bases (3).
Enseguida, otra técnica fue publicada por Frederick Sanger y colaboradores en el mismo año. Esta fue la técnica empleada por los siguientes 30 años. Se trata de un método enzimático con el cual se pueden secuenciar unos 900 pares de bases.
Mediante la incorporación de mejoras en los aspectos metodológicos y tecnológicos, el método Sanger permitió que el Proyecto Genoma Humano sea una realidad, secuenciando pequeños fragmentos de ADN humano que luego se ensamblarían. Frederick Sanger recibió su segundo premio Nobel en química por esta innovadora técnica.
A pesar de que en la actualidad ya existen nuevas metodologías más rápidas y menos costosas para el secuenciamiento, el método Sanger todavía se usa ampliamente para estudiar fragmentos de ADN.
Las técnicas de Maxam y Gilbert y la de Sanger son consideradas técnicas de secuenciamiento de primera generación. Desde aproximadamente una década, los investigadores cuentan con técnicas denominadas de nueva generación o más comúnmente conocidos como Next Generation Sequencing (NGS).
Las grandes diferencias entre una NGS y secuenciación de primera generación son la capacidad de procesar muchos fragmentos de ADN a la vez (hablamos de millones de fragmentos), la mayor rentabilidad y mucha más rapidez que los de primera generación. Además, se cuenta con diferentes plataformas para NGS.
En gran parte, el avance que ha tenido la tecnología de NGS corresponde al poder de la bioinformática. Con la bioinformática, se facilitaron el análisis y la manipulación de conjuntos de datos muy grandes, a menudo en el rango de gigabases (1 gigabase = 1,000,000,000 pares de bases de ADN).
El proyecto de secuenciación del genoma del SARS-CoV-2 circulante en el territorio paraguayo contempla la utilización de la tecnología de secuenciación de próxima generación (NGS).
De esta manera, como habíamos señalado, el proyecto servirá no solo para obtener información sobre las variantes circulantes en el Paraguay, sino que además permitirá que más científicos paraguayos accedan a utilizar nuevas tecnologías de secuenciación y equipará laboratorios que en el futuro seguirán usando NGS para otros proyectos.
Papel de la secuenciación de ADN en la pandemia
El mundo enfrenta la devastadora pandemia de COVID-19 causada por el beta-coronavirus SARS-CoV-2, y el campo de la “epidemiología genómica” ha ganado un papel indispensable en estudio de esta enfermedad.
Para vigilar la evolución de los microorganismos, la epidemiología genómica analiza los cambios que ocurren en el genoma de estos.
Las pruebas de amplificación de ácidos nucleicos (RT-PCR) han mostrado ser herramientas de gran valor para la detección rápida y precisa de infecciones por SARS-CoV-2. Sin embargo, poseen un uso limitado para identificar eventos de transmisión y detección de variantes circulantes.
La secuencia del ARN del virus SARS-CoV-2, causante de la pandemia de COVID-19, tiene pocos sitios variables en su genoma, esto es, sitios en donde se producen mutaciones, por lo que es posible seguir su evolución analizando esos sitios variables (4). Es aquí donde la secuenciación del material genético del virus cobra su importancia.
La acumulación de cambios de significancia biológica en el genoma del virus fue detectada casi en tiempo real, a medida que el virus se dispersaba por todo el mundo.
Gracias a esto y al conocimiento detallado del genoma, se logró el desarrollo y el mejoramiento de los métodos de diagnóstico moleculares y serológicos, así como la identificación de variantes que pudieran influir en el comportamiento de la pandemia, por ejemplo, en la tasa de transmisión o en la virulencia.
Además, la información sobre las variaciones genéticas del virus es una pieza fundamental en el desarrollo de vacunas y tratamientos. En particular, si hay cambios estructurales importantes (a consecuencia de esas mutaciones genéticas), puede ser necesario reevaluar la eficacia de los tratamientos e inmunizaciones disponibles.
Resultados de la secuenciación masiva de muestras de SARS-CoV-2
Científicos de todo el mundo secuencian el genoma completo de virus SARS-CoV-2 para la detección de las variaciones que el virus va presentando en el correr del tiempo desde su aparición. De esta manera es posible actuar precozmente en la implementación de protocolos sanitarios a nivel local, además de aportar los hallazgos de los investigadores al conocimiento científico sobre la enfermedad.
Mediante la secuenciación del material genético, tres variantes de importancia epidemiológica fueron detectadas en los últimos meses: la variante detectada en el Reino Unido, la detectada en pacientes de Sudáfrica y la detectada en Brasil.
El Reino Unido es uno de los países que realiza masivamente la secuenciación de las muestras positivas de SARS-CoV-2. Para este fin, varias universidades e institutos han formado un consorcio, que tiene como objetivo realizar la secuenciación del genoma completo de muestras del virus a gran escala y proveer ágilmente de información relevante a los centros locales del Sistema Nacional de Salud (NHS) y al gobierno del Reino Unido.
Sus datos ayudan a las agencias de salud pública a gestionar el brote de COVID-19 e informan los esfuerzos de investigación de vacunas.
Solo en el Reino Unido se lleva a cabo el 40 % de la secuenciación de genoma completo de SARS-CoV-2 de todo el mundo. Gracias a este singular esfuerzo, el Reino Unido detectó una nueva variante denominada B.1.1.7, que se destacó debido a una cantidad elevada de mutaciones.
Esta variante se propaga con mayor facilidad y rapidez, aunque continúan los estudios para saber si realmente es más letal o no. La variante se detectó por primera vez en setiembre del 2020 y rápidamente pasó a registrar una alta prevalencia en Londres y el sudeste de Inglaterra.
Desde entonces, se la ha detectado en numerosos países en todo el mundo, incluidos los Estados Unidos y Canadá (5).
Otra de las variantes, que fue detectada en octubre de 2020, es la que ha aparecido en Sudáfrica y es denominada B.1.351, la cual comparte algunas mutaciones con la variante detectada en el Reino Unido. También se han registrado casos provocados por esta variante fuera de Sudáfrica.
La variante detectada en pacientes de Brasil ha sido denominada B.1.1.28.1. La misma contiene una serie de mutaciones adicionales que podrían afectar su capacidad de ser reconocida por los anticuerpos (6).
Los ejemplos arriba citados de experiencias en países en los que se realiza la secuenciación del genoma completo nos muestran claramente la enorme importancia de que el Paraguay también realice este tipo de vigilancia.
Pues al momento no sabemos siquiera qué variantes son prevalentes en el país y esto puede tener gran importancia a la hora de evaluar las medidas sanitarias que deban adoptarse en determinado momento así como las opciones de tratamientos y vacunas más adecuadas para la población general.
Agradecimientos especiales a la Dra. Magalí Martínez por la colaboración en la revisión del texto.
Referencias
- Investigadores analizarán la genética del virus SARS-COV-2. Consejo Nacional de Ciencia y Tecnología (CONACYT).
- National Human Genome Research Institute (2019). DNA Sequencing Fact Sheet.
- Maxam AM, Gilbert W. A new method for sequencing DNA. Proc Natl Acad Sci U S A. 1977;74(2):560-564.
- Uso de la secuenciación genómica para visualizar la trazabilidad de la pandemia de COVID-19. Instituto Aragonés de Ciencias de la Salud.
- Variantes nuevas del virus que causa la COVID-19. (2021). Centers for Disease Control and Prevention (CDC).
-
Emerging SARS-CoV-2 Variants (2021). Centers for Disease Control and Prevention (CDC)
¿Qué te pareció este artículo?
 (5 votos, promedio: 5,00 de 5)
(5 votos, promedio: 5,00 de 5)Columnista y editora científica de Ciencia del Sur. MSc y PhD en Biología Parasitaria con énfasis en Biología Molecular aplicada a microorganismos por el Instituto Osvaldo Cruz (Fiocruz) de Río de Janeiro, Brasil. Fabiola obtuvo su licenciatura en Biología de la Facultad de Ciencias Naturales y Exactas de la Universidad Nacional de Asunción.
Realizó un posdoctorado en la Universidad de Bath (Inglaterra) y es colaboradora externa del Centro para el Desarrollo de la Investigación Cientifica. Fue Research Assistant en el Instituto Sanger de Cambridge. Actualmente, es Senior research technician in NGS. Departamento de Oncología de la Universidad de Cambridge, Inglaterra.
- Fabiola Román Maldonado

- Fabiola Román Maldonado

- Fabiola Román Maldonado

- Fabiola Román Maldonado

- Fabiola Román Maldonado

- Fabiola Román Maldonado

- Fabiola Román Maldonado

- Fabiola Román Maldonado
- Fabiola Román Maldonado
- Fabiola Román Maldonado
- Fabiola Román Maldonado
- Fabiola Román Maldonado
- Fabiola Román Maldonado

- Fabiola Román Maldonado

- Fabiola Román Maldonado
- Fabiola Román Maldonado

- Fabiola Román Maldonado

- Fabiola Román Maldonado
- Fabiola Román Maldonado

- Fabiola Román Maldonado

- Fabiola Román Maldonado

- Fabiola Román Maldonado

- Fabiola Román Maldonado

- Fabiola Román Maldonado

- Fabiola Román Maldonado

- Fabiola Román Maldonado

- Fabiola Román Maldonado

- Fabiola Román Maldonado

- Fabiola Román Maldonado

- Fabiola Román Maldonado
- Fabiola Román Maldonado









